90 % des patients atteints de HoFH présentent des variants du gène LDLR associés à la maladie1


Écoutez cette vidéo de 2 min sur le mécanisme de la HoFH
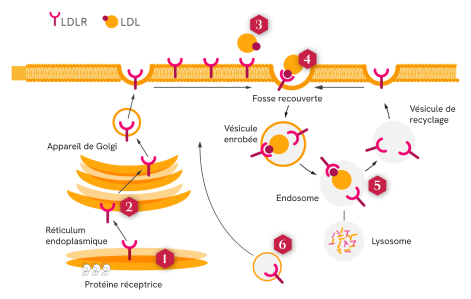
La HoFH est associée à des variants de plusieurs gènes affectant l’activité du récepteur des LDL (LDLR), eux-mêmes associés à la maladie2,3
La majorité des LDL est éliminée du plasma par des LDLR situés sur les membranes cellulaires des cellules hépatiques3
Ces variants associés à la maladie perturbent différents aspects fonctionnels des LDLR, qui peuvent être classés en six catégories3 :
 Absence de synthèse du précurseur protéique des LDLR
Absence de synthèse du précurseur protéique des LDLR Transport défectueux des LDLR dans le réticulum endoplasmique
Transport défectueux des LDLR dans le réticulum endoplasmique Liaison défectueuse aux LDL
Liaison défectueuse aux LDL Pas d’internalisation des LDLR ou des LDL à cause d’une agrégation défectueuse dans les puits de clathrine
Pas d’internalisation des LDLR ou des LDL à cause d’une agrégation défectueuse dans les puits de clathrine Aucun recyclage des LDLR
Aucun recyclage des LDLR Échec du transport des LDLR jusqu’à la surface des membranes cellulaires
Échec du transport des LDLR jusqu’à la surface des membranes cellulaires
Des variants d’autres gènes associés à la maladie perturbant la fonction des LDLR ont également été identifiés chez les patients atteints de HoFH3 :
- Mutations d’APOB qui altèrent la capacité de liaison d’Apo B aux LDLR
- Mutations gain de fonction de PCSK9 qui inhibent la fonction des LDLR et accélèrent leur dégradation
- Mutations de LDLRAP1 qui perturbent l’internalisation des LDLR et causent une forme rare d’HF, l’hypercholestérolémie autosomique récessive (ARH)* .
Apo B : apolipoprotéine B; LDLR : récepteur des lipoprotéines de basse densité; PCSK9 : proprotéine convertase subtilisine/kexine de type 9; LDLRAP1 : protéine adaptatrice du récepteur des lipoprotéines de basse densité de type 1; HF : hypercholestérolémie familiale; ARH : hypercholestérolémie autosomique récessive.
*Comme ces mutations sont récessives, les parents porteurs du gène défectueux peuvent présenter une cholestérolémie normale
Les variants associés à la maladie altèrent le fonctionnement de la voie des LDL et réduisent leur élimination dans le sang, ce qui entraîne une athérosclérose prématurée et des complications cardiovasculaires1,3,4
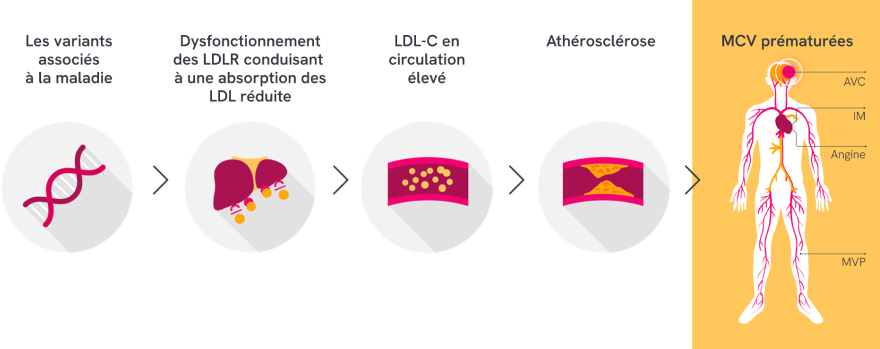
Cette figure s’applique à l’HF en général
Les variants du gène LDLR associés à la maladie causent une perte de fonction partielle (LDLR-défectueux) ou complète (LDLR-négatif) des LDLR2. Ces variants peuvent donner lieu aux génotypes suivants :
- Double LDLR-défectueux : les variants associés à la maladie entraînant une perte de fonction partielle sont présents sur les deux allèles.
- Double LDLR-négatif (nul/nul) : les variants associés à la maladie entraînant une perte de fonction complète sont présents sur les deux allèles
- LDLR-défectueux + LDLR-négatif
Les variants LDLR-négatifs associés à la maladie sont également associés à l’absence ou la quasi-absence de liaison et d’absorption des LDL3
Par rapport aux patients doublement LDLR-défectueux et LDLR-défectueux + LDLR-négatifs, les patients doublement LDLR-négatifs (nul/nul) présentent les caractéristiques suivantes3,5-7 :
- Taux de LDL-C supérieur
- Incidence des MCV supérieure
- Pronostic moins favorable
- Réponse réduite aux traitements pharmacologiques
La majorité des LDL est éliminée du plasma par des LDLR situés sur les membranes cellulaires hépatiques2
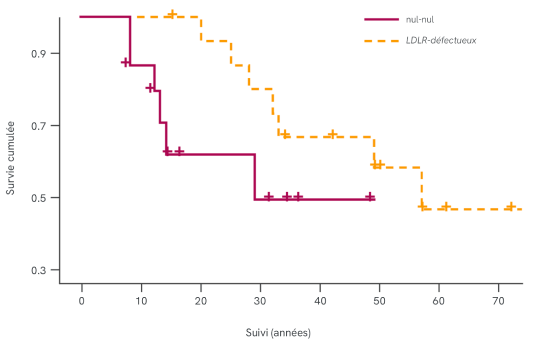
* Le registre SAFEHEART est un registre national de patients atteints d’HF vivant en Espagne. Les patients sont inscrits et suivis chaque année pour consigner les changements pertinents relatifs aux traitements hypolipidémiants et à l’apparition d’événements cardiovasculaires. Le graphique illustre les données de survie de 34 patients atteints d'HoFH inscrits entre 2004 et 2015.
Références
1. France, M., et coll. Atherosclerosis, 2016;255:128–139; 2. Cuchel, M., et coll. Eur Heart J, 2014;35:2146–2157; 3. Gidding, S.S., et coll. Circulation, 2015;132:2167–2192; 4. Bruckert, E. Atheroscler Suppl, 2014;15:26–32; 5. Alonso, R., et coll. J Clin Lipidol, 2016;10:953–961; 6. Sjouke, B., et coll. Eur Heart J, 2015;36:560–565.