Livelli estremamente elevati di colesterolo “cattivo” (LDL-C) possono esporre a rischio di eventi cardiovascolari?

Immagini non rappresentative di pazienti reali
Che cos'è l'HoFH?
L'HoFH è una malattia grave, ultra-rara e che dura tutta la vita, che si eredita dai genitori e significa che il corpo non è in grado di eliminare il colesterolo cattivo (LDL-C). Ciò può causare depositi di placca nelle arterie, determinando l aterosclerosi, e spesso porta ad attacchi cardiaci precoci, ictus e altre malattie che coinvolgono le arterie.1
L'HoFH viene in genere gestita con più approcci terapeutici, tra cui cambiamenti nello stile di vita, farmaci e altre terapie.1
Avere l'HoFH significa avere livelli estremamente elevati di colesterolo "cattivo" (LDL-C), che possono essere anche oltre 5 volte superiori al target (>10 mmol/L con HoFH vs target di 1,8 mmol/L per gli adulti senza rischio cardiovascolare).1
L'HoFH colpisce circa 1 persona su 320,000 in Italia, molte delle quali non vengono diagnosticate o vengono diagnosticate più avanti nella vita, il che ritarda il trattamento critico2
In che modo l'HoFH è
diverso dall'HeFH?
HoFH e HeFH possono sembrare abbastanza simili; infatti, un numero significativo di persone riceve inizialmente una diagnosi errata, poiché l'HoFH viene spesso confusa con l'HeFH.4
| FH omozigote (HoFH) | FH eterozigote (HeFH) |
|---|---|
| Geni mutati ereditati da entrambi i genitori | Gene mutato ereditato da un genitore |
| Si verifica in 1 persona su 300,000 | Si verifica in 1 persona su 250 |
| LDL-C >10 mmol/L (non trattato) |
Colesterolo LDL-C >4.1 mmol/L nei bambini; Colesterolo LDL-C >4.9 mmol/L negli adulti |
| Probabili sintomi fisici, come depositi di colesterolo negli occhi, nei tendini, nelle ginocchia, nei gomiti e/o tra le dita delle mani e dei piedi; non sempre i depositi sono presenti | Possibili sintomi fisici, come depositi di colesterolo negli occhi, nei tendini, nelle ginocchia, nei gomiti e/o tra le dita delle mani e dei piedi |
| Trattamento al momento della diagnosi, indipendentemente dall'età | Trattamento già all'età di 10 anni |
Adattato da McGowan e Cuchel et al.
Chi colpisce?
L'HoFH si manifesta quando si ereditano due copie mutate degli alleli del gene che causa l'FH, una da ciascun genitore; quei genitori molto probabilmente hanno la più comune FH eterozigote.4
La corretta identificazione dell'HoFH può interessare intere famiglie, poiché l'FH può essere individuata anche nei genitori e in altri parenti. Una persona con HoFH può avere fratelli e sorelle con HeFH.2
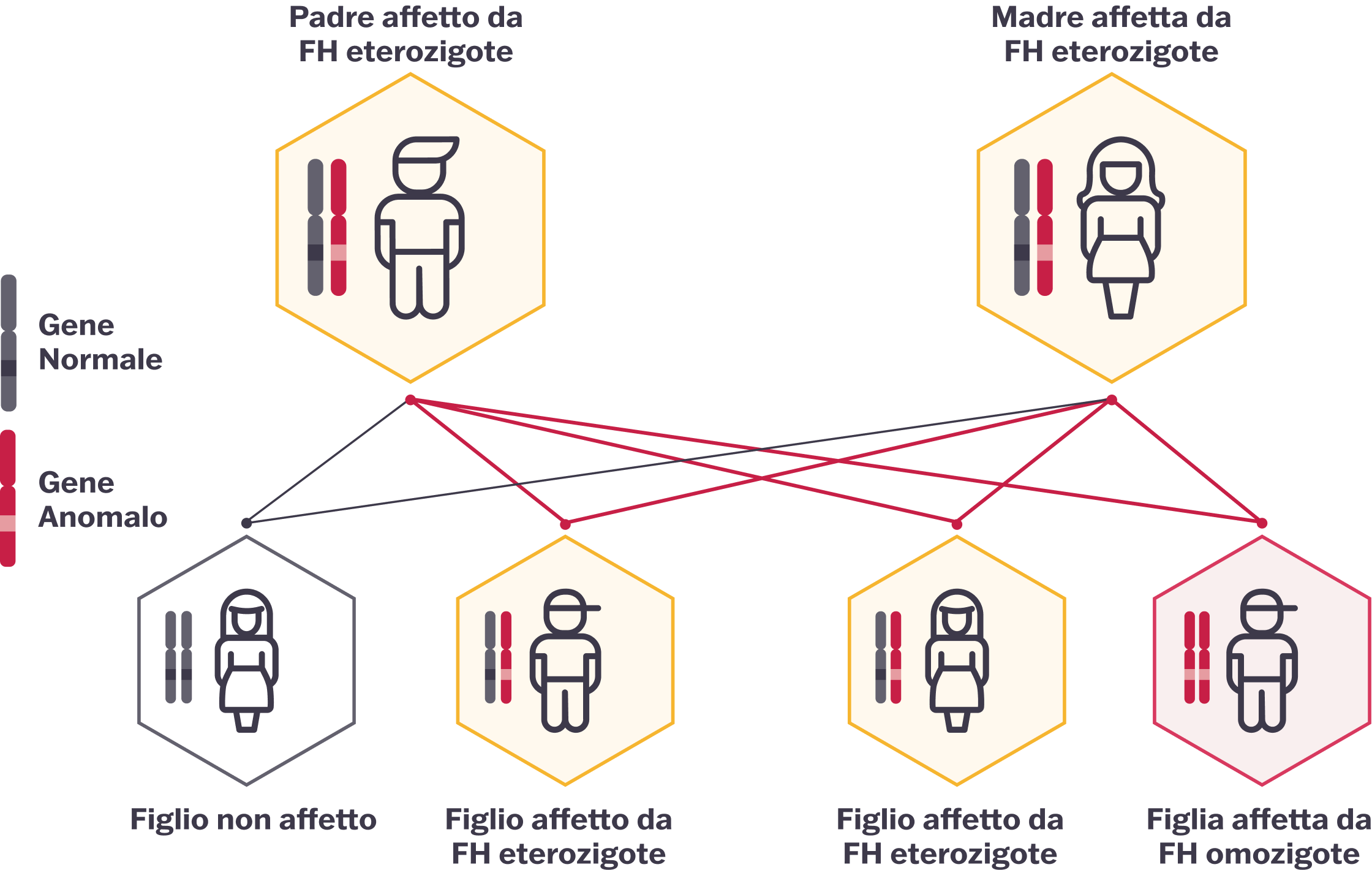
Adattato da Cuchel et al
Quali sono i rischi?
L'HoFH può progredire rapidamente e avere effetti per tutta la vita, che portano ad attacchi cardiaci prematuri, ictus e altri eventi cardiovascolari, spesso entro i primi due decenni.1
Una diagnosi mancata, una diagnosi errata o un trattamento non ottimale possono consentire alla malattia di progredire.4
Alla nascita
Livelli estremamente elevati di colesterolo "cattivo" (LDL-C fino a 13 mmol/L in un nascituro e 18 mmol/L in un neonato)4,5
Molti casi non vengono diagnosticati durante l'infanzia, quando la progressione della malattia può essere ancora prevenuta
Durante l'infanzia
I depositi di colesterolo si sviluppano nella pelle, nei tendini e nei vasi sanguigni4
Il colesterolo si accumula nelle valvole cardiache, causando problemi cardiaci4
Possibile attacco cardiaco prima dei 10 anni6,7
I pazienti con fenotipi gravi possono avere un infarto miocardico prima del decimo anno di età
Durante l'adolescenza
Primo infarto, ictus o altro evento cardiaco4
Continuano a svilupparsi depositi di colesterolo e calcio4
Il peggioramento dell'accumulo di tessuto fibroso e l'infiammazione possono portare a difetti cardiaci4
Molte persone con HoFH non vengono diagnosticate correttamente fino a quando non diventano adulte, il che ritarda il trattamento critico del loro LDL-C elevato4
Nell'adulto
Depositi di placca nelle arterie e malattia progressiva delle arterie4
Problemi alle valvole cardiache4
Insufficienza cardiaca, arresto improvviso del cuore (arresto cardiaco)4
Se non trattate, le persone con HoFH raramente superano i 30 anni4

Impara i segnali

Immagini non rappresentative di pazienti reali
Altri membri della famiglia hanno ipercolesterolemia familiare (può essere l'HoFH o la più comune HeFH)1,3
Livelli estremamente elevati di colesterolo "cattivo" (LDL-C >10 mmol/L senza trattamento)1
Eventi cardiovascolari precoci, come infarto o ictus9
Segni visibili, come l'accumulo di grasso sotto la pelle (xantomi), che compaiono prima dei 10 anni4
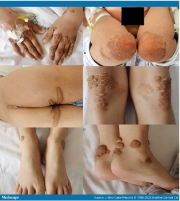
Esempi di
xantomi visti
nei bambini8
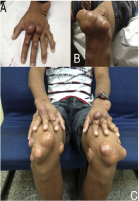
Esempi di
xantomi visti
negli adulti10
ASCVD, disturbo cardiovascolare aterosclerotico; CVD, disturbo cardiovascolare; IM, infarto del miocardio
Se si sopetta l'HoFH, contattare il proprio medico, che fornirà tutte le informazioni necessarie
L'HoFH deve essere trattata immediatamente da uno specialista (come un cardiologo o un endocrinologo)
Trova un centro specializzato nelle vicinanzeFONTI BIBLIOGRAFICHE
1. Cuchel M et al. Eur Heart J. 2023;44(25):2277-2291. 2. Tromp TR et al. Lancet. 2022;399:719-728. 4. Cuchel M et al. Eur Heart J. 2014;35:2146-2157. 5. de Gennes JL et al. Arteriosclerosis. 1985;5:440-442. 6. Raal FJ et al. Atherosclerosis. 2018;277:483-492. 7. Al-Shaikh AM et al. Cardiol Young. 2002;12:105-112. 3. McGowan MP et al. J Am Heart Assoc. 2019;8:e013225. 9. Santos R et al. Lancet. 2016;4(10):850-861. 8. Wang N et al. J Med Case Rep. 2022;16(1):290. 10. Rocha VZ et al. J Am Coll Cardiol. 2013;61(21):2193.